Yasutomi Higashikuni, M.D., Ph.D., FESC
Assistant Professor of Cardiovascular and Genetic Research
Jichi Medical University
Biography
Dr. Yasutomi Higashikuni is an Assistant Professor of Cardiovascular and Genetic Research at Jichi Medical University. His research interests include homeostatic inflammation, RNA metabolism and modification, aging, and synthetic biology in cardiovascular medicine. He is leading a new research field, “Synthetic Cardiology”, which creates novel gene and cell therapies that enable automatic reversal of aging phenotypes and maintenance of homeostasis.
- Synthetic Cardiology
- Homeostatic Inflammation
- RNA Metabolism and Modification
- Aging
Ph.D. in Internal Medicine, 2010
The University of Tokyo
M.D., 2002
The University of Tokyo
Experience
Research interests include, but not limited to:
- Synthetic Cardiology
- RNA metabolism and modification
- Homeostatic Inflammation
- Aging
Research interests include, but not limited to:
- Synthetic Cardiology
- RNA metabolism and modification
- Homeostatic Inflammation
- Aging
Research interests include, but not limited to:
- Synthetic Cardiology
- RNA metabolism and modification
- Homeostatic Inflammation
Tim Lu Lab:
- Living Cell Computation and Memory
- Development of Novel Synthetic Antimicrobial Peptide
- Combinatorial Phenotypic Screening
- Next-Generation Gene Therapy for Heart Failure
Higashikuni/Sata Lab:
- Homeostatic Inflammation in Cardiovascular Disease (Including Multi-Organ Interaction)
- Anti-Oxidant Response in Cardiovascular Disease
- Renin-Angiotensin System in Cardiovascular Disease
Clinical Activity:
- Percutaneous Coronary Intervention
Higashikuni/Sata Lab:
- Homeostatic Inflammation in Cardiovascular Disease (Including Multi-Organ Interaction)
- Anti-Oxidant Response in Cardiovascular Disease
- Renin-Angiotensin System in Cardiovascular Disease
Clinical Activity:
- Percutaneous Coronary Intervention
Sata Lab:
- Homeostatic Inflammation in Cardiovascular Disease (Including Multi-Organ Interaction)
- Anti-Oxidant Response in Cardiovascular Disease
- Renin-Angiotensin System in Cardiovascular Disease
Clinical Activity:
- Percutaneous Coronary Intervention
Accomplishments
Projects

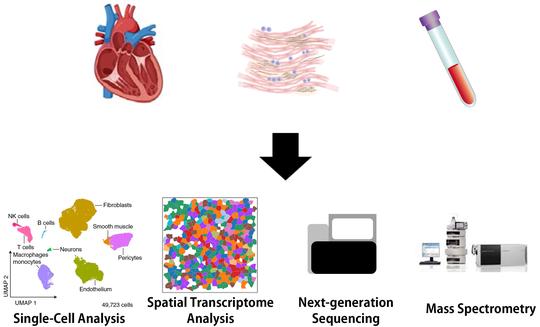



Recent Posts

Recent & Upcoming Talks
People
Meet the Team
Principal Investigator
Yasutomi Higashikuni, M.D., Ph.D., FESC
Assistant Professor of Cardiovascular and Genetic Research
Researchers
Yu Tanaka, M.D., Ph.D.
Assistant Professor of Pediatrics
Wenhao Liu, M.D.
Graduate Student
Takumi Obana
Technical Assistant
Featured Publications
Recent Publications
Contact
- higashikuni.yasutomi@jichi.ac.jp; yasutomihigashikuni@g.ecc.u-tokyo.ac.jp
- +81-285-58-7341 (Ext. 3101)
- 3311-1 Yakushiji, Education & Research Center, Shimotsuke-shi, Tochigi 329-0498
- Enter Education & Research Center and take the stairs or the elevator to Division of Cardiovascular and Genetic Research at Center for Molecular Medicine on Floor 4